Alkenes: Addition & Oxidation Reactions
Forward: General Mechanistic Traits of Alkene Addition Reactions







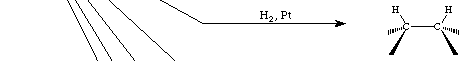







General Mechanisms of Alkene Addition Reactions:
The majority of the reactions of alkenes which will be described in this section fall into three basic categories:
- ionic additions, which are initiated by an electrophilic agent interacting with the alkene p-cloud, activating the alkene carbons to nucleophilic additions,
- syn addition reactions occurring on one side of the alkene p-cloud, by either radical or concerted mechanisms, and,
- oxidative cleavage reactions in which the carbon-carbon double bond is cleaved to form di-carbonyl derivatives.
Syn additions to the alkene p-cloud which are covered in this section include hydrogenation, gem-diol formation from MnO4-, organoborane formation from BH3 and carbene-dependent cyclopropanation reactions. In each of these, an electrophilic agent reacts with the p-system to (more or less) simultaneously form bonds to both carbons. In hydrogenation, the hydrogen gas is rendered electrophilic by adsorption to a metal surface (i.e., Pt). The metal surface also binds the alkene, activating the addition, and a variety of carbon-metal intermediate species are probably involved. Trivalent boron is a powerful Lewis acid and reaction with BH3 is probably initiated by the formation of a p-complex, as described above; this complex, however, seems to decompose in a concerted fashion to form the syn borane.
Oxidative cleavage of alkene generally involves the intermediate formation of a bridged oxygen species of some sort, followed by spontaneous or reductive cleavage. The examples covered in this section include ozonolysis, where the intermediate ozonide is decomposed by a dissolving-metal reduction, and acid-catalyzed cleavage of the intermediate metal di-ester formed during MnO4- oxidation.
As you work through each of these sets of reactions, you should pay particular attention to the common features of the mechanisms in each group. An understanding of a few basic chemical generalities will help you view reactions as broad classes, and help you avoid overt memorization.


The resulting carbocation is formed on the carbon of the alkene which is best able to stabilize the cationic center. In simple unstrained non-conjugated systems, without adjacent heteroatoms, the order of stability of carbocations will be tertiary > secondary > primary. Since tertiary centers have no attached hydrogens, secondary centers have one and primary centers have two, there is an apparent inverse relationship between the "number of attached hydrogens" and the likelihood that the carbocation will form at that center. This is the origin of Markovnikov's Rule, which states that...
often restated as "them that has, gets". While the rule is a useful guide, you should remember that the selectivity is actually to place the carbocation on the carbon which can best stabilize the charge.
Once the carbocation is formed, the most favorable reaction will involve the addition of a nucleophile to form an sp3 center. In the reaction with halogen acid (HCl and HBr), the most nucleophilic molecules in the system will be the chloride and bromide anions. Attack of these on the planar (sp2) carbocation can occur from either above, or below the plane defined by the sp2 center, and the net addition of HX can therefore occur either syn (cis; on the same side) or anti (trans; on the opposite side), relative to the hydrogen atom.



The bromine radical is electrophilic and attacks the alkene p-system, which donates an electron, forming a s>-bond to the bromine and leaving an unpaired electron (a radical) on one of the carbons of the alkene. If the alkene is asymmetrical, bond formation can occur by two pathways, as described above for halogen acid addition. Once again, the reaction pathways leading to the two radicals will differ in energy, and, in general, the pathway leading to the more stable intermediate will be of lower energy, and will be the preferred pathway.
The resulting radical is formed on the carbon of the alkene which is best able to stabilize the electrophilic site (the unpaired electron). In simple unstrained non-conjugated systems, without adjacent heteroatoms, the order of stability of carbon radicals parallels that of carbocations, with tertiary > secondary > primary. Since tertiary centers have no attached hydrogens, secondary centers have one and primary centers have two, there is an apparent inverse relationship between the "number of attached hydrogens" and the likelihood that the radical will form at that center. The carbon radical which is formed abstracts a hydrogen atom (most likely from HBr), propagating the chain and giving one mole of product. In this product, the "hydrogen" attached to the center which formed the radical, that is, the center with "the fewest number of hydrogens" (a secondary or tertiary center) and the bromine is attached to the carbon which is adjacent to the most stable radical ("the center with the most hydrogens"). This is opposite to Markovnikov's Rule, as described in the previous example, and the orientation in this reaction is often termed "anti-Markovnikov". While the rule is a useful guide, you should remember that the selectivity is actually to place the radical character on the carbon which can best stabilize the unpaired electron (the electrophilic center).
As with carbocation intermediates, carbon radicals are planar (sp2), and hydrogen abstraction from the second molecule of HBr can occur from either above, or below the plane defined by the sp2 center. The net addition of HBr can therefore occur either syn (cis; on the same side) or anti (trans; on the opposite side), as described in the previous example.
The addition of halogen to alkenes is a stepwise process involving a "halonium" ion intermediate. The formation of this intermediate is initiated through attack of halogen on the alkene p-system, to form the cyclic halonium ion (i.e., bromonium or chloronium ion) and expel the halogen anion (i.e., bromide or chloride). This intermediate is highly electrophilic and reacts rapidly with the best nucleophile in the system; that is, the halide anion expelled in the previous step. Since the halonium ion effectively blocks attack by halide on the same side, attack must come from the backside (relative to the large halogen atom) to form the trans-1,2-dihalide. This is demonstrated below for the addition of bromine to 1-propene. The large bromine on the intermediate bromonium ion (shown as a space-filling overlay) effectively blocks attack from the top, forcing the addition to be anti (trans; from the opposite side). The attack of Br+ on the bromonium ion is an example of an SN2 reaction in which a nucleophile attacks at the carbon and displaces the leaving group is a single, smooth, concerted process..


As before, the large halogen on the intermediate halonium ion effectively blocks attack from the top, forcing the addition of halogen and hydroxide to be anti (trans; on the opposite side), relative to each other.

Once the carbocation is formed, reaction with water (a relatively poor nucleophile) will be sufficiently slow such that the carbocation has a long enough lifetime to undergo rearrangement. In general, the molecule will rearrange by a combination of alkyl-, methyl- and hydride-transfers to form the most stable carbocation. Because of this, acid-catalyzed hydration can only be utilized to prepare alcohols where rearrangement is not considered to be a problem (i.e., tertiary and some secondary alcohols).

The reaction of alkenes with mercuric acetate follows the general mechanism for Lewis acid activation of alkene addition reactions. Loss of acetate anion and chelation of the mercury with the alkene p-cloud generates a bridged, cationic intermediate. As in the previous examples, if the alkene is asymmetrical, the more positive charge will be localized on the carbon of the alkene which is best able to stabilize it (tertiary > secondary > primary). The steric bulk of the bridged mercury ion will direct attack to the opposite face of the alkene and the best nucleophile in the system (hydroxide anion) will attack at the carbon bearing the greatest positive charge. Again, this follows the Markovnikov convention.

The reaction of BH3 with an alkene begins with the Lewis acid chelation of the alkene p-system by the boron. This complex then rearranges in a more or less concerted manner to produce the alkyl borane, as shown below. The reaction seems to be dominated by steric effects and the boron attaches to the least hindered carbon. All three equivalents of the boron hydride can be utilized in separate reactions to give a trialkyl borane.



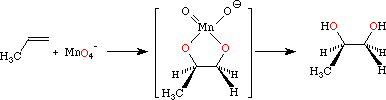
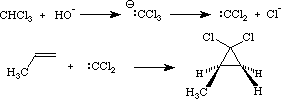

Oxidation of Alkenes
Ozonolysis of alkenes, followed by dissolving metal reduction with Zn dust in acid, smoothly converts alkenes into aldehydes and ketones, depending on the nature of the groups attached to the sp2 carbons. The reaction involves the intermediate formation of an ozonide, in which the two halves of the alkene are joined by bridging oxygen atoms. Reduction the ozonide forms aldehydes from terminal alkenes and from sp2 centers attached to one additional carbon. Likewise, ketones are formed from sp2 centers attached to two additional carbons.
